我们是专注于大健康品牌全案策划设计的专业机构。品牌战略,CIS导入,品牌定位,产品策划,品牌设计,包装设计,整合推广......
我们提供大健康一站式定制化解决方案,提升产品力和品牌力,为品牌营销赋能升级!
医药制药、医疗器械、健康食品、保健食品、有机农业、消毒防疫、美妆护理......
十多年做好一件事,因为专注,所以专业!
我们提供大健康一站式定制化解决方案,提升产品力和品牌力,为品牌营销赋能升级!
医药制药、医疗器械、健康食品、保健食品、有机农业、消毒防疫、美妆护理......
十多年做好一件事,因为专注,所以专业!

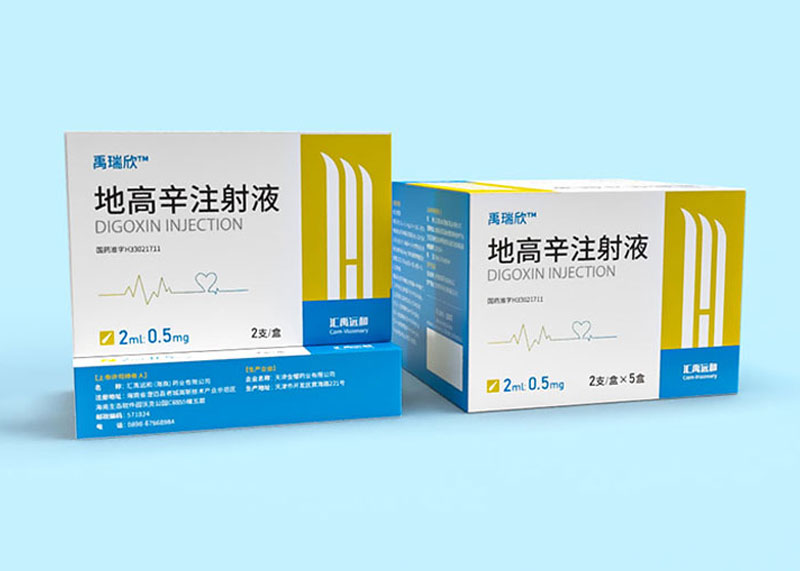
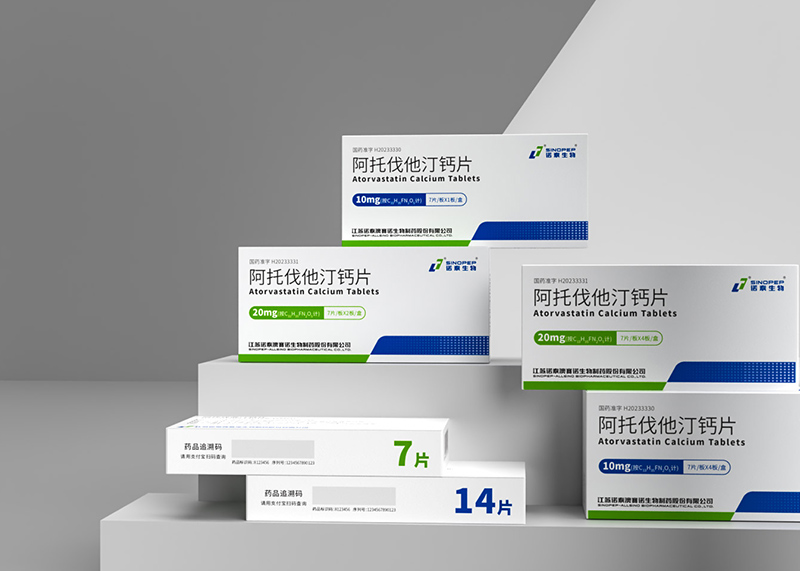
产品命名、产品定位、产品卖点、系列规划、单品爆款打造、包装设计、外观专利注册,印刷制作、终端规范
产品命名、产品定位、产品卖点、系列规划、单品爆款打造、包装设计、外观专利注册,印刷制作、终端规范
产品命名、产品定位、产品卖点、系列规划、单品爆款打造、包装设计、外观专利注册,印刷制作、终端规范
产品命名、产品定位、产品卖点、系列规划、单品爆款打造、包装设计、外观专利注册,印刷制作、终端规范
产品命名、产品定位、产品卖点、系列规划、单品爆款打造、包装设计、外观专利注册,印刷制作、终端规范
15年来,石特大健康以医药健康品牌全案服务立足,是专业的医药品牌策划公司、医药品牌设计公司、药品包装设计公司以及医药品牌宣传公司。
服务过华润三九、太极药业、远大健康、华海药业、康恩贝等一大批医药上市公司及大健康企业,
为中药/化药、创新药/仿制药、品牌OTC、生物制剂、医疗器械、保健品/滋补品、特医食品、健康食品等
打造具有销售力的药品包装、品牌/产品形象及宣传推广服务,一站式解决医药健康企业品牌力、产品力、市场力问题。
15年来,石特大健康以医药健康品牌全案服务立足,是专业的医药品牌策划公司、医药品牌设计公司、药品包装设计公司以及医药品牌宣传公司。

中国生物

华润三九

华东医药

雷允上

太极绵药

康恩贝

华海药业

远大健康

人福医药

佐力药业

奥吉娜药业

康正药业

海正药业

臣功制药

森科制药

华康股份

京新药业

中邦制药

迪安诊断

九洲药业

远力健药业

江北药业

黄河药业

济民可信

康亚药业

核力欣健

视方极

仙琚药业

威海华洋药业

爱诺药业

欣乐加生物

可帮基因

赛美视

火山石医药

澎尚医药

亚瑟医药

上药九洲

心创医疗

恩瑞特

亚州生物

瑞莱谱医疗

强生医疗中国

术创医疗

安迪斯医疗

汇禹远和
15年深耕大健康领域,打造核心“三板斧”
大健康产业研究+全员考核培训,成员平均服务年限5年以上





项目周期要多久?
根据项目实际情况,创作周期是不定的,一般情况下,石特会依据项目情况和节点提供排期,同客户商议,并严格执行推进,正常情况下都会在合同约定时间内提交方案。
如果设计不满意怎么办?
石特以客户满意为根本,对已签约客户,原则上,我们不限修改次数。经过前期的充分沟通和细致的策划调研,呈现出来的设计往往都是适合的方案。在项目过程中,团队会认真听取客户意见,力求给客户最满意的作品。
石特提供印刷制作服务吗?
应客户和市场需求,石特提供一站式设计、印刷、制作服务,设计完成后如希望委托石特制作的,请与我们项目经理联系,我司会安排设计师看样跟色,以达到最佳的成品效果!
石特可以免费设计样稿吗?
设计是一项系统性工作,简单样稿并不能准确反映需求和水平,同时为了尊重设计师的劳动成果,除正规招标外,原则上我司不提供免费设计服务。
石特收费情况是怎样的?
石特力争为客户提供高性价比的服务,会站在客户的角度最大程度的节省成本,这也是很多客户选择与我们长期合作的一个重要原因。另外,选择石特年度服务的客户,在此基础上,我们会有更大力度的优惠和更全面的配套服务,具体来说,品牌服务是定制化服务,价格依据需求而定!
石特团队服务怎么样?
区别于传统广告公司,石特独创1+1+1+N模式,每个项目不论大小,我们都有专门的项目经理全程对接,策划和设计总监全程把关,并根据项目实际情况配备相应创作成员,力争达到最佳产出效果,让客户省心放心!

项目基本情况了解,电话/视频/当面沟通。

合同签订,时间规划,板块细分。

项目组成立,内部讨论,市场调研洞察,核心策略方向。

品牌定位策略,品牌传播策略,品牌营销策略。

品牌策划整体方案,视觉设计方案,整合传播方案。

品牌传播执行,中间测试改良。

效果评估,项目完成。

 0571-8868558715605889708
0571-8868558715605889708